Download the original slides for this lecture here
Presenters: Alyssa Klein, Wei Zhao
Introduction to Available Computing Clusters
We will first begin by discussing two computing clusters at NIH: CCAD and Biowulf. As this is a course from DCEG, we will open this section with a brief description of the Computer Cluster at DCEG (CCAD). CCAD is a cluster only for DCEG members. This is mostly optional information for interested DCEG members as all following practical and lecture sessions will focus on Biowulf when cluster use is required.
Computer Cluster at DCEG (CCAD)
CCAD is the dedicated computing cluster for DCEG. CCAD operates under a fair use policy to avoid monopolization of resources. In other words, users are given equal access to the available resources as they become available.
For example, say the job queue is empty and User A submits 8 jobs to the cluster which can run 5 jobs at any time. The first 5 of User A’s jobs are then started while the other 3 remain in the queue. Meanwhile User B submits 1 job which is put in the queue after User A’s 3 remaining jobs. When one of User A’s jobs completes, User B’s job will then be run despite being entered into the queue after User’s A’s to ensure fair use. Job scheduling and fair use assurance is managed automatically using the Sun Grid Engine management software, through which all jobs must be submitted.
The two types of cluster use include interactive sessions and cluster jobs.

Interactive sessions allow for actions to be performed on the command line after logging into the cluster. Logging onto CCAD logs a user onto a specialized login node which provides a place for interactive use but does not actively control the CCAD cluster. Cluster jobs are the primary and preferred method of using CCAD in which users submit a job to a queueing system to be run when resources become available, according to fair use.
For your reference, here are the resources available at CCAD:
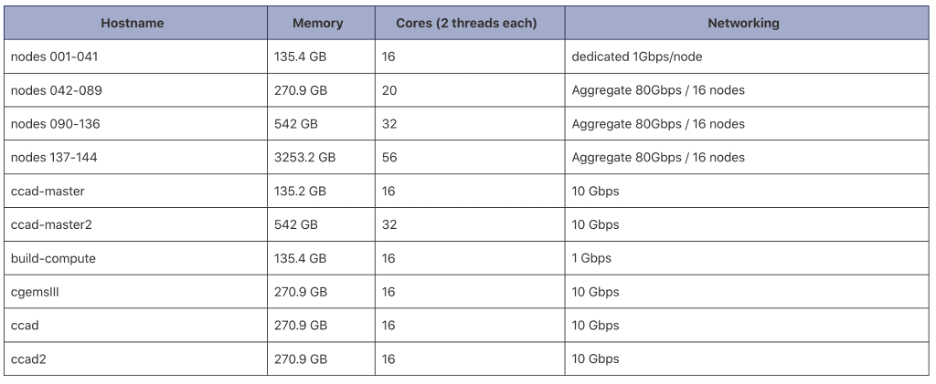
Additional CCAD Resources
-
Additional CCAD information can be found at myDCEG:
Biowulf
As compared to CCAD, Biowulf is a much larger computer cluster available to all of NIH.
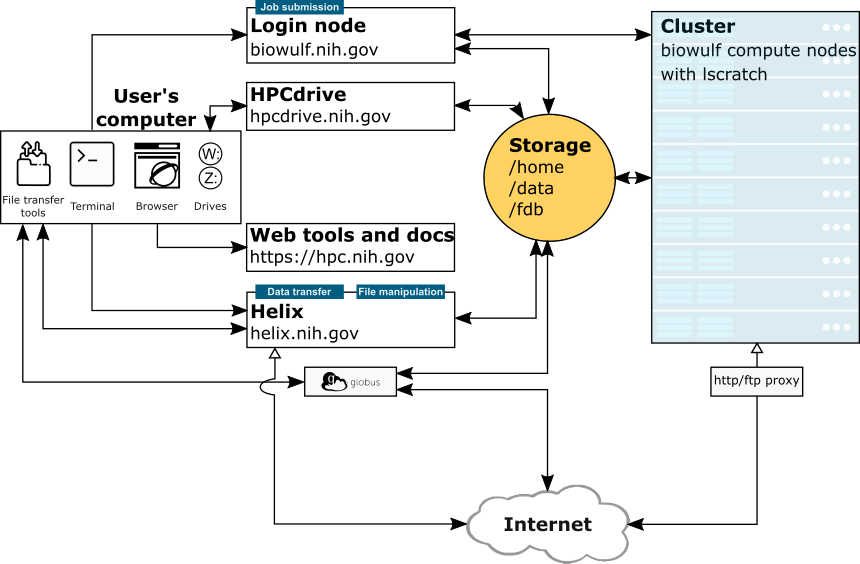 from https://hpc.nih.gov/systems/
from https://hpc.nih.gov/systems/
Like CCAD, Biowulf also operates under a fair use policy by which jobs are prioritized according to each user’s recent usage volume, measured in CPU-hours. If cluster resources are in high demand, users with lower recent usage (measured in CPU-hours) have their jobs prioritized over users with high recent usage. Jobs are scheduled automatically using a workload management software called Slurm, analogous to the Sun Grid Engine noted for CCAD.
Note that, like CCAD, there is a login node in on the cluster. This login node is what you are operating immediately when you login to Biowulf, and it is shared among all users. For this reason, scientific applications and other computation-intensive processes must be run via job submission or others will be unable to use the cluster.
Biowulf is one of the top 500 most powerful clusters in the world, featuring:
- 100,000+ cores
- 200,000+ CPUs
- 4,000+ nodes
- 900+ TB memory
- 3+ PB local scratch (lscratch)
- ~40 PB high performance storage
- 5 PB object storage
- 800+ GPUs (4,000,000+ CUDA cores)
Biowulf offers many versions of over 1000 scientific applications, and maintains data from several public databases for user convenience such as reference genomes, NCBI nt Blast database, NCBI taxonomy database, ClinVar, gnomAD, etc. Biowulf also features many versions of both python and R with over 500 and 1600 packages installed, respectively, and supports containerization using Singularity.
For information on using Biowulf, see the Biowulf website which contains tons of great information. In particular, we want to highlight the in-depth education and training resources on everything from basic linux to advanced scripting for Biowulf, and also see the detailed guides for using individual scientific softwares.
Additional Biowulf Resources
-
Submit help tickets: send email to staff@hpc.nih.gov
Comparison of Biowulf and CCAD
Here is a table to summarise and highlight the differences between working with CCAD and Biowulf:
| Biowulf | CCAD | |
|---|---|---|
| Job submission | sbatch --cpus-per-task=# --mem=#g --job-name #JobName --time ##:##:## myscript.sh |
module load sge |
| Interactive jobs | sinteractive --cpus-per-task=#cpus --mem=#g |
module load sge |
| Cancel jobs | scancel #job-id |
qdel job_id [options] |
| Monitor jobs | squeue, sjobs, jobload, jobhist |
qstat |
| Transfer data | Globus, WinSCP/Fugu | scp, rsync |
| Load applications | module spider #module |
module load #module |
Snapshots
Snapshots are a read-only copy of data at a particular point in time. These snapshots are very helpful if you inadvertently delete a file that you need. To locate the file you are interested in, you can go to a specific snapshot by using the following command:
cd .snapshot
This will take you to a main snapshot directory that contains all of the other snapshot directories. From there you can find the file you need and copy it back to the desired directory.
Here are the policies of both clusters on snapshots:
| Biowulf | CCAD |
|---|---|
| - Home dir: Weekly backups, with daily incremental backups - Data dir: NOT BACKED UP - Buy-in storage - Additional information: File Backups and Snapshots on the HPC Systems |
- Nightly snapshots last one week - 6 hour snapshots last 3 days - True backups done via CBIIT taken weekly and retained based on their policies - Permanent backups need to be requested to be transferred to the archive |
More information on Biowulf snapshots can be found here: https://hpc.nih.gov/storage/backups.html.
Cluster How-Tos: Connect, Transfer Files/Share Data
CCAD and Biowulf are primarily accessed for one of two purposes: direct use of the cluster or transferring files. Here we will summarise how to do both.
| Host | Hostname | Accessible by | Purpose |
|---|---|---|---|
| Biowulf | biowulf.nih.gov | All HPC users | cluster head node |
| Helix | helix.nih.gov | All HPC users | data transfer |
| HPCdrive | hpcdrive.nih.gov | All HPC users | data transfer |
| CCAD | ccad.nci.nih.gov | All HPC users | cluster head node |
| CCAD Tdrive | gigantor.nci.nih.gov | All HPC users | data transfer |
Connecting via SSH
The only method for directly accessing either cluster is ultimately through the command line. This is done via secure shell, or SSH.
Connecting via SSH will vary depending on whether you’re using a MacOS or Windows computer to connect. Windows users will need to install PuTTY to connect via SSH.

If you are using a Mac, no additional software is necessary. Simply open the Mac app Terminal and type ssh -Y [user]@biowulf.nih.gov, where [user] is your login ID.
Note: -Y enables trusted X11 forwarding. X11 forwarding allows for the use of graphical applications on a remote server (ex. Integrative Genomics Viewer- IGV). Another option for running graphics applications is NoMachine (NX) (https://hpc.nih.gov/docs/nx.html#setup).
We will detail and practice connecting to CCAD and Biowulf further in the practical section.
Transfer of files can be accomplished one of many ways:
Graphical User Interface (GUI) file transfer applications
GUI-based transfer applications can be a convenient way to transfer data. WinSCP for Windows and FileZilla for both Windows and MacOS are free applications recommended for file transfers.


Mounting drives
Drives from the NIH HPC and CCAD can be mounted directly to your local computer which allows you to click and drag files in familiar fashion. This is best only for small file transfers; transfer of larger files should be done through another method.

Doing so will require you to specify a folder to be mounted. Refer to the table below for the correct path formatting.
| Description | Directory at cluster | SMB path for Windows | SMB path for Mac | |
|---|---|---|---|---|
| Biowulf/Helix | user’s home directory | /home/[user] | \\hpcdrive.nih.gov\[user] | smb://hpcdrive.nih.gov/[user] |
| data directory | /data/[user] | \\hpcdrive.nih.gov\data | smb://hpcdrive.nih.gov/data | |
| user’s scratch space | /scratch/[user] directory | \\hpcdrive.nih.gov\scratch\[user] | smb://hpcdrive.nih.gov/scratch/ [user] |
|
| shared group area (e.g. you are a member of group PQRlab) | /data/PQRlab | \\hpcdrive.nih.gov\PQRlab | smb://hpcdrive.nih.gov/PQRlab | |
| CCAD | main directory | /home/ | \\gigantor.nci.nih.gov\ifs | smb://gigantor.nci.nih.gov/ifs |
| user’s home directory | /home/[user] | \\gigantor.nci.nih.gov\ifs \DCEG\Home\[user] |
smb://gigantor.nci.nih.gov/ifs /DCEG/Home/[user] |
. For more detailed instructions on how to mount drives on Biowulf, see here.
Globus file transfer
Globus is the recommended method to transfer and share files from Biowulf. Globus has the ability to monitor transfer performance, retry failures, recover from faults automatically, and report transfer status. See here for how to set up a Globus account.

Command line file transfer
Finally one can use the command line to transfer files using the secure FTP (Windows via PuTTY) and secure copy (Windows via PuTTY, MacOS) commands, like so to transfer to the HPC:
Windows via PuTTY:
pscp source_folder/my_file_1.txt username@helix.nih.gov:/destination_folderMacOS:
scp source_folder/my_file_1.txt username@helix.nih.gov:/destination_folder
Or conversely from the HPC:
Windows via PuTTY:
pscp username@helix.nih.gov:/source_folder/my_file_2.txt destination_folderMacOS:
scp username@helix.nih.gov:/source_folder/my_file_2.txt destination_folder
. Note that these commands use the Helix system, not Biowulf (as designated by @helix.nih.gov). Biowulf should not be used for file transfers unless done from within a job on the cluster.
Basic Linux Commands
Linux Programming
Because CCAD and Biowulf must be accessed via the command line it is necessary to know some linux before using either. We’ve already discussed a few command line programs such as ssh for connecting to the clusters and scp for file transfer, but more is required to operate them. We will cover some of the most useful commands in the practical section for this session.
For now, to learn more about linux please see this linux cheatsheet and vim cheatsheet we’ve prepared for your reference, and/or view these external resources:
- Biowulf’s ‘Introduction to Linux’
- Biowulf’s ‘Introduction to Bash Scripting’
- Biowulf’s online classes for Bash
- Biowulf’s Linux and Slurm cheatsheet
- Vim documentation
- Cheatsheet for Vim
Bioinformatics File Formats and Tools
Finally we will discuss some of the most important file formats used in bioinformatics. These file formats will include the ones you are likely to encounter in a typical genomics study, but there are still many more specialized file formats which we won’t cover in this session.
Before we describe these file formats in more detail, below is a workflow diagram tracing the flow of information over the course of a cancer study and a slightly more detailed description of each format in the table below.
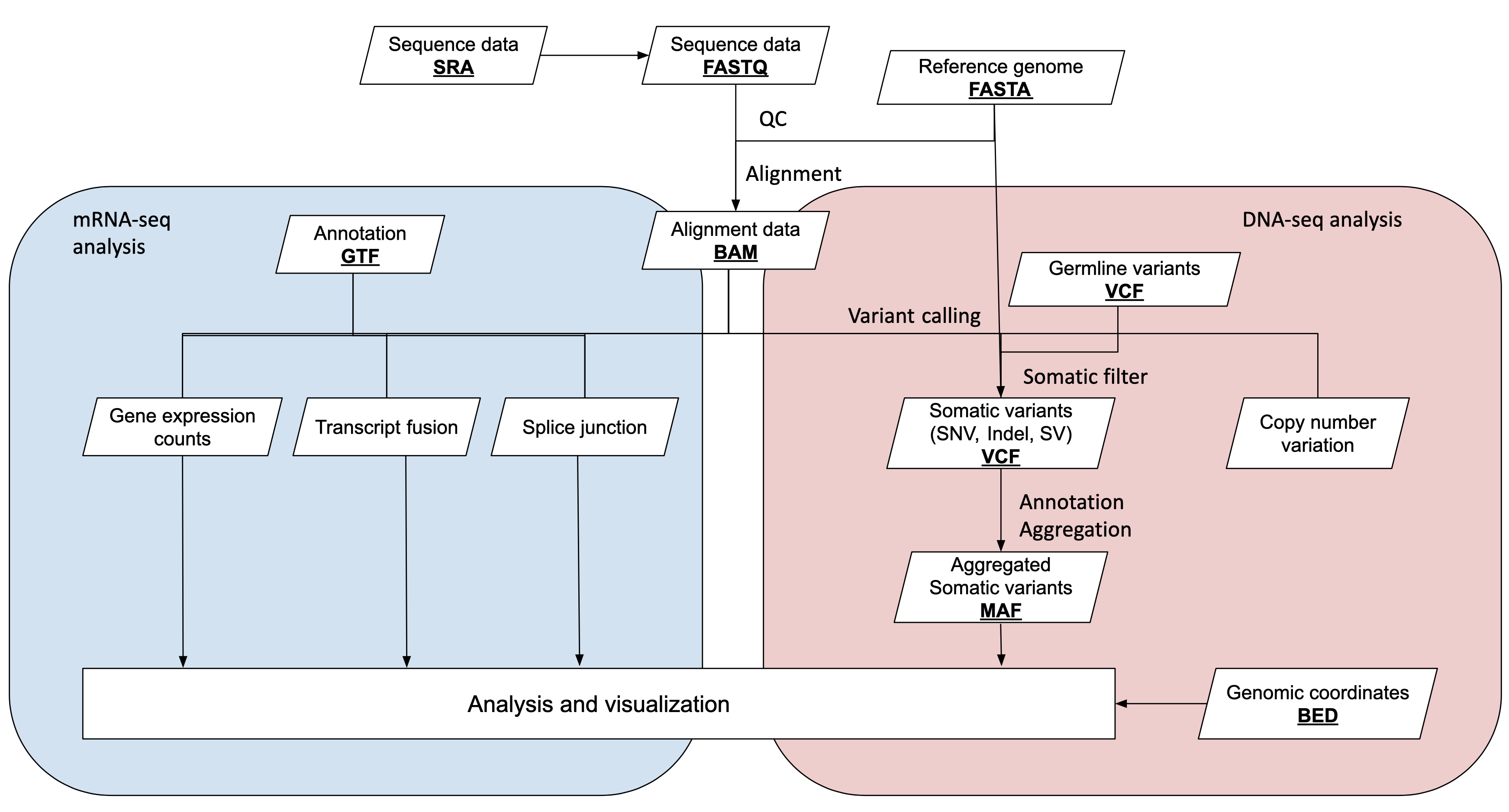
Raw Data
Sequencing data with per-base quality score is stored in one of two forms: fastq (raw reads), SAM/BAM/CRAM (aligned reads).
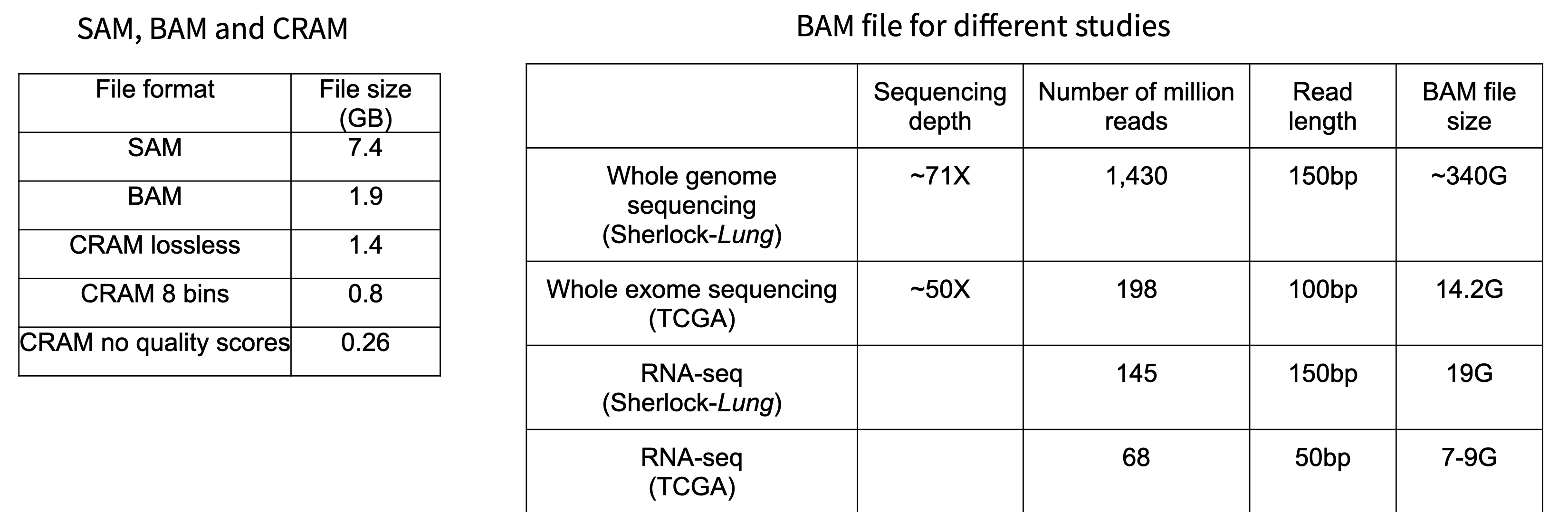
Fastq is the most basic sequencing format and stores only sequence barcodes, sequenced bases, and per-base quality scores. 1 nucleotide base in fastq format is stored in ~2 bytes (sequence + quality score). Bearing in mind that the human genome itself is 3GBs, one 30x whole genome sequencing sample= 3GB * 30x * 2 bytes per base ~ 180GB. Compressing fastq files typically compresses to 25% original size.
Fastq sequencing files which are aligned to reference genomes are stored as alignment files in SAM/BAM/CRAM format. SAM (Sequence Alignment Map) is a human-readable, uncompressed alignment file, whereas BAM and CRAM are both compressed forms of SAM. BAM/CRAM files are considerably smaller than SAM files and are therefore the most common alignment formats for data sharing and deposit.
BAM is a lossless compression; this means that the original SAM is always perfectly reconstructed during decompression. CRAM offers a greater degree of compression from BAM, and can be either lossless or lossy (i.e. some information may be lost during decompression).
Processed Data
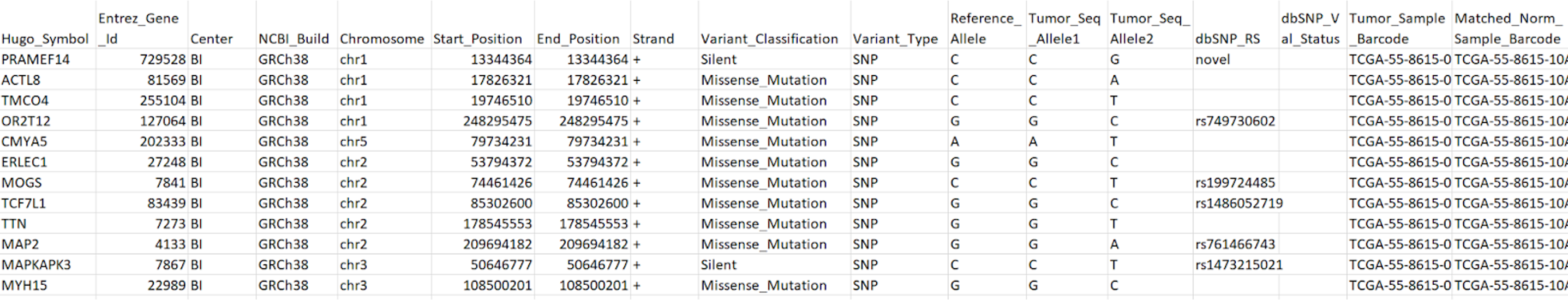
Variants calling results can be shared in the formats of VCF or MAF. In somatic genomic studies, each VCF file is generated for one tumor/normal pair. In germline studies it on VCF contains pooled variants for all samples.

MAF files contain aggregated mutation information from VCF files and are generated on a project-level. MAF files also include annotation of variants from public databases.
Preparing Data for Database Submission
- NCBI Sequence Read Archive (SRA)
- requires raw data with per-base quality scores for all submitted data.
- accepts binary files such as BAM/CRAM, HDF5 (for PacBio, Nanopore), SFF (when BAM is not available, for 454 Life Science and Ion Torrent data), and text formats such as FASTQ.
- For more information: https://www.ncbi.nlm.nih.gov/sra/docs/submit/
- Gene Expression Omnibus (GEO)
- studies concerning quantitative gene expression, gene regulation, epigenetics, or other functional genomic studies. (e.g. mRNA-seq, miRNA-seq, ChIP-Seq, HiC-seq, methyl-seq/bisulfite-seq)
- does NOT accept WGS, WES, metagenomic sequencing, or variation or copy number projects.
- a complete submission includes: metadata, processed data, raw data containing sequence reads and quality scores (will be submitted to SRA by the GEO team)
- For more information: https://www.ncbi.nlm.nih.gov/geo/info/seq.html
- The database of Genotypes and Phenotypes (dbGaP)
- studies investigating the interaction of genotype and phenotype in humans.
- all submissions that require controlled access must be submitted through dbGaP.
- requires registration of the study and subjects prior to data submission.
- raw data will be submitted to the protected SRA account.
- For more information: https://www.ncbi.nlm.nih.gov/sra/docs/submitdbgap/
File formats
Finally, we present a summary table of the bioinformatics file formats and what tools are available to work with them:
| Format name | Data type | Tools |
|---|---|---|
| SRA | a raw data archive with per-base quality score | sra-tools |
| FASTA | a text file of reference genome sequence data | FASTA Tools |
| FASTQ | a text file of sequencing data with quality score | FastQC, FASTX-Toolkit, Seqtk, Samtools, Picard tools |
| SAM/BAM/CRAM | formats of sequence alignment data | Samtools, Picard tools |
| BCF/VCF/gVCF | a tab-delimited text file to store the variation calls | bcftools |
| BED (PEBED) | a tab-delimited text file to store the coordinates of genomic regions. | bedtools |
| GTF/GFF/GFF3 | a tab-delimited text file to describe genes or other features | gff tools, GFF utilities (gffread, gffcompare) |
| MAF | a tab-delimited text file with aggregated mutation information from VCF files and are generated on a project-level. | MAFtools |
Additional Resources
For full descriptions of aforementioned formats as well as tools to work with them, see the resources below as well as the supplementary information tab for this session.